Analyze StrainGE output in Python
Now that we have run StrainGST and StrainGR (including the compare step), how do we analyze the outputs? This page uses Python and its commonly used data science stack (NumPy, SciPy, Pandas and matplotlib+seaborn) to parse the data, plot the relative abundances of strains over time, and generate an ACNI/gap similarity plot.
Download data
We download an archive containing StrainGE outputs part of the vignette described in the paper on the persistence of an E. coli strain in the gut of a woman with recurrent urinary tract infections. The extracted data is organized in a straingst and straingr folder.
[2]:
!curl --output umb_data.tar.gz https://raw.githubusercontent.com/broadinstitute/strainge-paper/master/umb/umb_data.tar.gz
!tar -xzvf umb_data.tar.gz
% Total % Received % Xferd Average Speed Time Time Time Current
Dload Upload Total Spent Left Speed
100 12196 100 12196 0 0 42347 0 --:--:-- --:--:-- --:--:-- 42347
x straingst/UMB11_01.tsv
x straingst/UMB11_02.tsv
x straingst/UMB11_03.1.tsv
x straingst/UMB11_03.tsv
x straingst/UMB11_04.1.tsv
x straingst/UMB11_04.tsv
x straingst/UMB11_05.tsv
x straingst/UMB11_06.tsv
x straingst/UMB11_07.tsv
x straingst/UMB11_08.tsv
x straingst/UMB11_11.tsv
x straingst/UMB11_12.tsv
x straingr/UMB11_01.tsv
x straingr/UMB11_02.tsv
x straingr/UMB11_03.1.tsv
x straingr/UMB11_03.tsv
x straingr/UMB11_04.1.tsv
x straingr/UMB11_04.tsv
x straingr/UMB11_05.tsv
x straingr/UMB11_06.tsv
x straingr/UMB11_07.tsv
x straingr/UMB11_08.tsv
x straingr/UMB11_11.tsv
x straingr/UMB11_12.tsv
x straingr/compare.summary.chrom.txt
Import required modules
[3]:
from pathlib import Path
import numpy
import pandas
import matplotlib.pyplot as plt
from IPython.display import display
StrainGST
Read StrainGST outputs and combine it in a DataFrame
The TSV files written by StrainGST contain both sample statistics (the first two lines), and statistics for each identified strain (see StrainGST page). In this tutorial, we are mainly interested in the identified strains. In the code below, when calling pandas.read_csv, we give the argument skiprows=2 to skip the sample statistics.
[16]:
STRAINGST_DIR = Path("straingst/")
df_list = []
sample_names = []
for f in STRAINGST_DIR.glob("*.tsv"):
sample_name = f.stem
df = pandas.read_csv(f, sep='\t', comment='#', skiprows=2, index_col=1)
df_list.append(df)
sample_names.append(sample_name)
# Combine all StrainGST results from each sample into a single DataFrame.
straingst_df = pandas.concat(df_list, keys=sample_names, names=["sample"])
sample_names = list(sorted(sample_names, key=lambda e: float(e.replace("UMB11_", ""))))
straingst_df.sort_index()
[16]:
| i | gkmers | ikmers | skmers | cov | kcov | gcov | acct | even | spec | rapct | old_rapct | wscore | score | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sample | strain | ||||||||||||||
| UMB11_01 | Esch_coli_NGF1 | 0 | 49631 | 49622 | 50090 | 0.985 | 7.009 | 6.831 | 0.980 | 0.987 | 1.000 | 1.536 | 1.505 | 0.940 | 0.940 |
| UMB11_02 | Esch_coli_NGF1 | 0 | 49631 | 49623 | 5358 | 0.103 | 1.546 | 0.158 | 0.932 | 0.707 | 1.032 | 0.048 | 0.045 | 0.047 | 0.048 |
| UMB11_03 | Esch_coli_1190 | 0 | 48261 | 48249 | 37144 | 0.711 | 2.814 | 1.975 | 0.926 | 0.826 | 0.972 | 0.395 | 0.366 | 0.437 | 0.449 |
| UMB11_03.1 | Esch_coli_1190 | 0 | 48261 | 48254 | 31201 | 0.595 | 2.152 | 1.264 | 0.918 | 0.830 | 1.030 | 0.711 | 0.652 | 0.365 | 0.376 |
| UMB11_04.1 | Esch_coli_1190 | 0 | 48261 | 48250 | 19042 | 0.362 | 1.870 | 0.668 | 0.908 | 0.743 | 1.047 | 0.135 | 0.122 | 0.173 | 0.182 |
| UMB11_05 | Esch_coli_1190 | 0 | 48261 | 48248 | 40411 | 0.775 | 2.741 | 2.097 | 0.923 | 0.884 | 0.967 | 0.484 | 0.447 | 0.540 | 0.558 |
| UMB11_06 | Esch_coli_1190 | 1 | 48261 | 21600 | 30714 | 0.794 | 7.102 | 5.565 | 0.283 | 0.797 | 0.552 | 1.005 | 0.391 | 0.079 | 0.142 |
| Esch_coli_H3 | 0 | 45610 | 45560 | 74449 | 0.960 | 114.343 | 109.038 | 0.921 | 0.960 | 0.976 | 16.420 | 16.042 | 0.795 | 0.814 | |
| UMB11_07 | Esch_coli_1190 | 0 | 48261 | 48237 | 58276 | 0.854 | 4.557 | 3.846 | 0.803 | 0.873 | 0.794 | 0.411 | 0.822 | 0.415 | 0.522 |
| Esch_coli_f974b26a-5e81-11e8-bf7f-3c4a9275d6c8 | 1 | 47727 | 21265 | 17074 | 0.441 | 2.588 | 1.077 | 0.526 | 0.668 | 1.012 | 0.613 | 0.106 | 0.102 | 0.103 | |
| UMB11_08 | Esch_coli_1190 | 0 | 48261 | 48248 | 31599 | 0.592 | 2.243 | 1.309 | 0.904 | 0.810 | 0.980 | 0.315 | 0.285 | 0.344 | 0.351 |
| UMB11_11 | Esch_coli_1190 | 0 | 48261 | 48248 | 49462 | 0.920 | 6.107 | 5.545 | 0.920 | 0.923 | 0.965 | 1.180 | 1.086 | 0.696 | 0.721 |
| UMB11_12 | Esch_coli_1190 | 0 | 48261 | 48235 | 66509 | 0.941 | 9.061 | 8.431 | 0.800 | 0.941 | 0.734 | 0.978 | 2.020 | 0.489 | 0.667 |
| Esch_coli_26561 | 1 | 46249 | 19738 | 21112 | 0.853 | 4.773 | 3.983 | 0.780 | 0.869 | 0.968 | 1.548 | 0.395 | 0.486 | 0.502 |
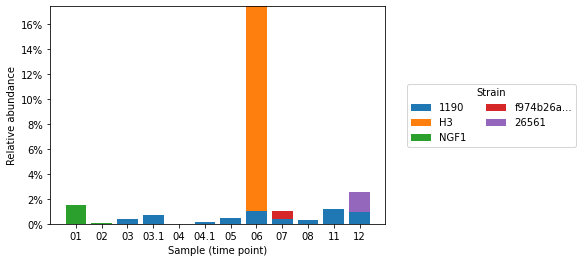
Plot relative abundances
[5]:
plt.figure(figsize=(6, 4))
strain_order = ['Esch_coli_1190', 'Esch_coli_H3', 'Esch_coli_NGF1', 'Esch_coli_f974b26a-5e81-11e8-bf7f-3c4a9275d6c8', "Esch_coli_26561"]
strain_labels = ['1190', 'H3', 'NGF1', 'f974b26a...', "26561"]
xlabels = [s.replace("UMB11_", "") for s in sample_names]
x = numpy.arange(len(sample_names))
bottom = numpy.zeros((len(sample_names),))
for ref, label in zip(strain_order, strain_labels):
# Create an array with all relative abundances for the current reference in each sample. If not available, set to zero.
rel_abun = numpy.array([
straingst_df.loc[(sample, ref), 'rapct'] if (sample, ref) in straingst_df.index else 0.0
for sample in sample_names
])
plt.bar(x, rel_abun, bottom=bottom, tick_label=xlabels, label=label, width=0.8)
bottom += rel_abun
plt.xlabel("Sample (time point)")
plt.ylabel("Relative abundance")
plt.gca().yaxis.set_major_formatter("{x:g}%")
plt.legend(title="Strain", loc="center left", bbox_to_anchor=(1.05, 0.5), ncol=2)
plt.show()
StrainGR
Load call data in a DataFrame
To load StrainGR outputs, we use a similar approach as descried above. In this case, the StrainGR TSV files can be directly loaded with pandas without skiprows.
One thing to note, StrainGR outputs metrics for every contig in the concatenated reference used for alignment. The output file thus contains metrics for contigs from strains that were not predicted to be present by StrainGST. We use the presence/absence predictions of StrainGST as our “truth” and remove the contigs from strains that weren’t present.
We apply a few other filters, including removing plasmid contigs, and contigs with less coverage than 0.5x.
[28]:
STRAINGR_DIR = Path("straingr/")
df_list = []
sample_names = []
for f in STRAINGR_DIR.glob("*.tsv"):
df = pandas.read_csv(f, sep='\t', index_col=0)
df = df.drop(index='TOTAL') # Remove TOTAL statistics
df_list.append(df)
sample_names.append(f.stem)
straingr_df = pandas.concat(df_list, keys=sample_names, names=["sample"])
straingr_df['straingst_present'] = straingr_df.index.map(lambda ix: ix in straingst_df.index)
straingr_df['is_plasmid'] = straingr_df['length'] < 4e6
straingr_df['enough_cov'] = straingr_df['coverage'] > 0.5
# Filter and re-index
straingr_df = straingr_df[straingr_df['straingst_present'] & ~straingr_df['is_plasmid'] & straingr_df['enough_cov']].reset_index().set_index(['sample', 'ref'])
straingr_df
[28]:
| name | length | coverage | uReads | abundance | median | callable | callablePct | confirmed | confirmedPct | ... | multiPct | lowmq | lowmqPct | high | highPct | gapCount | gapLength | straingst_present | is_plasmid | enough_cov | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sample | ref | |||||||||||||||||||||
| UMB11_11 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 2.492 | 106232 | 0.449 | 2 | 2819899 | 57.538 | 2818902 | 99.965 | ... | 0.004 | 435239 | 8.881 | 488 | 0.010 | 9 | 165998 | True | False | True |
| UMB11_06 | Esch_coli_H3 | NZ_CP010167.1 | 4630919 | 47.519 | 1331869 | 7.902 | 48 | 3863102 | 83.420 | 3861892 | 99.969 | ... | 0.004 | 1439048 | 31.075 | 84996 | 1.835 | 9 | 120001 | True | False | True |
| Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 1.963 | 69465 | 0.264 | 2 | 1515111 | 30.915 | 1513886 | 99.919 | ... | 0.015 | 1161951 | 23.709 | 699045 | 14.264 | 9 | 165099 | True | False | True | |
| UMB11_07 | Esch_coli_f974b26a-5e81-11e8-bf7f-3c4a9275d6c8 | NZ_LR536430.1 | 4975029 | 0.700 | 14686 | 0.132 | 0 | 378975 | 7.618 | 377743 | 99.675 | ... | 0.013 | 495996 | 9.970 | 17165 | 0.345 | 17 | 347002 | True | False | True |
| Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 1.591 | 59112 | 0.347 | 1 | 1894740 | 38.661 | 1893628 | 99.941 | ... | 0.010 | 323110 | 6.593 | 3384 | 0.069 | 12 | 171056 | True | False | True | |
| UMB11_03 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 0.822 | 35131 | 0.143 | 1 | 859998 | 17.548 | 859671 | 99.962 | ... | 0.001 | 160054 | 3.266 | 26 | 0.001 | 9 | 185085 | True | False | True |
| UMB11_01 | Esch_coli_NGF1 | NZ_CP016007.1 | 5026105 | 3.549 | 85824 | 0.823 | 3 | 2506998 | 49.880 | 2506921 | 99.997 | ... | 0.003 | 1668501 | 33.197 | 3681 | 0.073 | 1 | 16868 | True | False | True |
| UMB11_03.1 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 0.596 | 24708 | 0.278 | 0 | 547015 | 11.162 | 546816 | 99.964 | ... | 0.001 | 99001 | 2.020 | 114 | 0.002 | 7 | 172806 | True | False | True |
| UMB11_08 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 0.580 | 24145 | 0.118 | 0 | 505713 | 10.319 | 505494 | 99.957 | ... | 0.001 | 115822 | 2.363 | 64 | 0.001 | 6 | 158445 | True | False | True |
9 rows × 23 columns
Load compare data in a DataFrame
The above data mainly contains data per sample of individual strains as compared to its closest reference. In general, we are often more interested how strains in each sample relate to each other. These kind of relationships are computed with the straingr compare command. Here, we load the data from compare, make sure we only include comparisons between strains that were predicted to be present by StrainGST, and plot the ACNI/gap similarity.
[27]:
compare_df = pandas.read_csv(STRAINGR_DIR / "compare.summary.chrom.txt", sep='\t', index_col=[0, 1, 2])
def both_straingst_present(ix):
sample1, sample2, ref = ix
return (sample1, ref) in straingr_df.index and (sample2, ref) in straingr_df.index
compare_df['both_present'] = compare_df.index.map(both_straingst_present)
compare_df = compare_df[compare_df['both_present']].copy()
compare_df
[27]:
| scaffold | length | common | commonPct | single | singlePct | singleAgree | singleAgreePct | sharedAlleles | sharedAllelesPct | ... | BnotAweak | BnotAweakPct | Agaps | AsharedGaps | AgapPct | Bgaps | BsharedGaps | BgapPct | gapJaccardSim | both_present | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sample1 | sample2 | ref | |||||||||||||||||||||
| UMB11_03 | UMB11_03.1 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 126732 | 2.5859 | 126732 | 100.0000 | 126725 | 99.9945 | 126725 | 99.9945 | ... | 5 | 17.8571 | 185085 | 163905 | 88.5566 | 172806 | 172806 | 100.0000 | 0.9919 | True |
| UMB11_06 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 336010 | 6.8561 | 335954 | 99.9833 | 335809 | 99.9568 | 335865 | 99.9568 | ... | 160 | 57.3477 | 185085 | 163905 | 88.5566 | 165099 | 158136 | 95.7825 | 0.9895 | True | |
| UMB11_07 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 405110 | 8.2660 | 405067 | 99.9894 | 405023 | 99.9891 | 405066 | 99.9891 | ... | 65 | 32.5000 | 185085 | 175075 | 94.5917 | 171056 | 154895 | 90.5522 | 0.9872 | True | |
| UMB11_08 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 117635 | 2.4003 | 117635 | 100.0000 | 117629 | 99.9949 | 117629 | 99.9949 | ... | 2 | 3.8462 | 185085 | 145541 | 78.6347 | 158445 | 146928 | 92.7312 | 0.9971 | True | |
| UMB11_11 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 611275 | 12.4727 | 611224 | 99.9917 | 611203 | 99.9966 | 611254 | 99.9966 | ... | 29 | 11.9835 | 185085 | 185085 | 100.0000 | 165998 | 165998 | 100.0000 | 0.9889 | True | |
| UMB11_03.1 | UMB11_06 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 215863 | 4.4046 | 215823 | 99.9815 | 215758 | 99.9699 | 215798 | 99.9699 | ... | 95 | 71.4286 | 172806 | 172806 | 100.0000 | 165099 | 158136 | 95.7825 | 0.9922 | True |
| UMB11_07 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 252557 | 5.1533 | 252524 | 99.9869 | 252490 | 99.9865 | 252523 | 99.9865 | ... | 56 | 43.0769 | 172806 | 172806 | 100.0000 | 171056 | 148033 | 86.5407 | 0.9879 | True | |
| UMB11_08 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 75899 | 1.5487 | 75897 | 99.9974 | 75894 | 99.9960 | 75896 | 99.9960 | ... | 2 | 10.0000 | 172806 | 148091 | 85.6978 | 158445 | 146928 | 92.7312 | 0.9916 | True | |
| UMB11_11 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 390913 | 7.9764 | 390894 | 99.9951 | 390880 | 99.9964 | 390899 | 99.9964 | ... | 14 | 14.0000 | 172806 | 172806 | 100.0000 | 165998 | 154893 | 93.3102 | 0.9916 | True | |
| UMB11_06 | UMB11_07 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 718395 | 14.6585 | 718228 | 99.9768 | 717916 | 99.9566 | 718083 | 99.9566 | ... | 151 | 22.1083 | 165099 | 158136 | 95.7825 | 171056 | 148033 | 86.5407 | 0.9898 | True |
| UMB11_08 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 199668 | 4.0741 | 199626 | 99.9790 | 199556 | 99.9649 | 199598 | 99.9649 | ... | 18 | 11.1111 | 165099 | 151355 | 91.6753 | 158445 | 158445 | 100.0000 | 0.9951 | True | |
| UMB11_11 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 1059446 | 21.6174 | 1059249 | 99.9814 | 1058911 | 99.9681 | 1059108 | 99.9681 | ... | 50 | 6.2500 | 165099 | 158136 | 95.7825 | 165998 | 154893 | 93.3102 | 0.9964 | True | |
| UMB11_07 | UMB11_08 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 235328 | 4.8017 | 235291 | 99.9843 | 235271 | 99.9915 | 235308 | 99.9915 | ... | 6 | 5.1724 | 171056 | 131119 | 76.6527 | 158445 | 146928 | 92.7312 | 0.9909 | True |
| UMB11_11 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 1284089 | 26.2011 | 1283913 | 99.9863 | 1283763 | 99.9883 | 1283939 | 99.9883 | ... | 26 | 3.8981 | 171056 | 154895 | 90.5522 | 165998 | 160523 | 96.7018 | 0.9900 | True | |
| UMB11_08 | UMB11_11 | Esch_coli_1190 | NZ_CP023386.1 | 4900891 | 358765 | 7.3204 | 358752 | 99.9964 | 358744 | 99.9978 | 358757 | 99.9978 | ... | 5 | 3.6765 | 158445 | 146928 | 92.7312 | 165998 | 139943 | 84.3040 | 0.9945 | True |
15 rows × 32 columns
[41]:
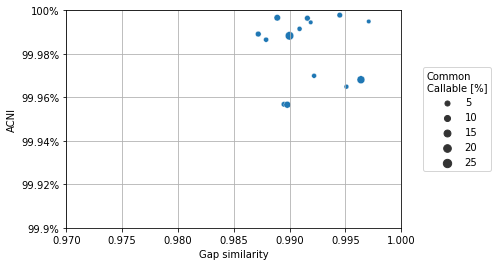
import seaborn
seaborn.scatterplot(x="gapJaccardSim", y="singleAgreePct", size="commonPct", data=compare_df)
plt.xlim(0.970, 1.0)
plt.xlabel("Gap similarity")
plt.ylim(99.9, 100)
plt.ylabel("ACNI")
plt.gca().yaxis.set_major_formatter("{x:g}%")
plt.grid('on')
plt.legend(title="Common\nCallable [%]", loc="center left", bbox_to_anchor=(1.05, 0.5))
[41]:
<matplotlib.legend.Legend at 0x160142700>